
Epigenomics
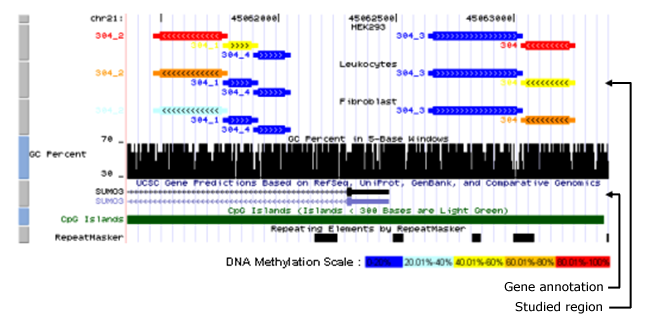
The goal of our epigenome projects funded through the national and international programs DEEP, NOTOX and BLUEPRINT is to gain a deeper understanding where, when and how DNA-methylation influences the regulation & function of our genome in cells of healthy and diseased states. Deciphering the epigenomes will allow us to read and understand the grammar and meaning of the epigenetic codings along human chromosomes. This information will be of great importance for many health related questions.
We perform state of the art epigenome analyses using HiSeq2500 and GSFLX technologies combined with Illumina array technologies. We develop comprehensive semi-automated experimental and bioinformatic workflows and tools for a comprehensive data interpretation. In addition we develop and apply novel high resolution bisulfite based sequencing protocols to ultimately analyse epigenomes at the single cell level. l allow us to follow and interpret changes in DNA-methylation on both DNA strands to understand mechanisms of de novo and de-methylation. We link our data on DNA-methylation dynamics to functional studies by investigating the structural effect of DNA-methylation on DNA function and stability. Moreover we investigate the processes that lead to the establishment of stable methylations patterns in cells and tissues in the context of cellular differentiation.
A large part of our epigenomic mapping work is currently funded by the BMBF funded program DEEP (2012-2017). DEEP (Deutsches Epigenom-Program) aims to decipher up to 100 Epigenomes of primary human cells at highest possible resolution and deposit them in public repositories as a recource for biomedical science. (see http://www.deutsches-epigenom-programm.de/research/). The DEEP program is part of the larger world wide International Human Epigenome Consortium IHEC.
In the DFG funded collaborative research initiative "Physical modeling of non-equilibrium processes in biological systems" SFB 1027 (see https://nestor.uni-saarland.de/fak7/rieger/sfb/home.html) we investigate the dynamic and structural aspects of DNA-methylation pattern formation using ultra deep sequencing approaches combined with hairpin-bisulfite sequencing.