
Applications
RNA Sequencing
In the field of RNA sequencing, a wide variety of questions find application. Both total RNA with a high proportion of ribosomal RNA and enriched forms such as mRNA, small (miRNAs) and non-coding RNAs can be sequenced using standardized protocols. In addition to sequencing whole cell assemblies or tissues, transcriptomes of single cells can now be generated. This contributes to the understanding of transcriptional heterogeneity between cells of the same type and between cells of different origin and function.
Please find more information addressing the method, biological questions and published articles here.
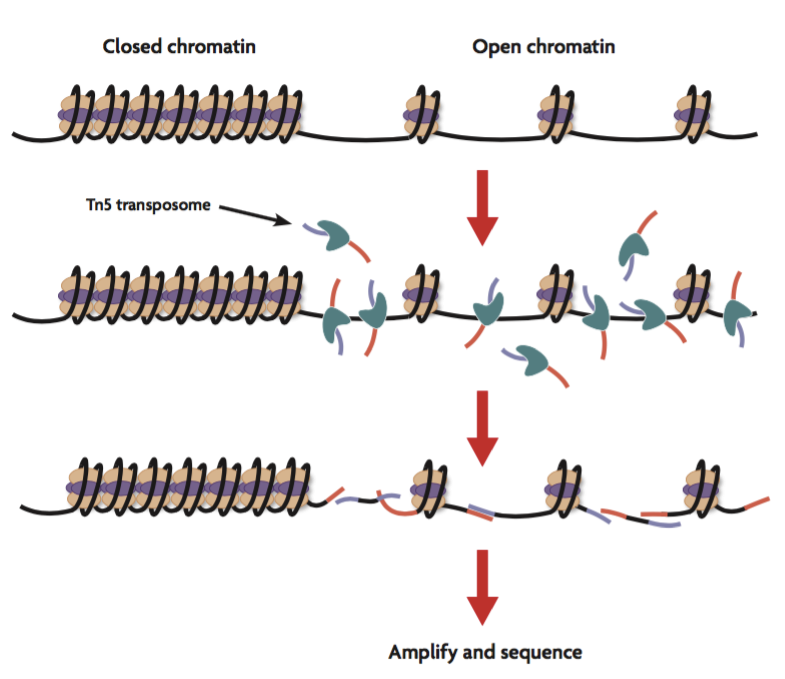
ATAC-seq/NOMe-seq
Chromatin structure has a major influence on genome integrity and the control of gene expression and is therefore a key element in cellular processes such as differentiation, aging and degeneration (e.g. cancer cell development and progression). ATAC-seq (Assay for Transposase-Accessible Chromatin sequencing) is a method for mapping open chromatin regions which have applications to whole cell assemblies or tissues as well as at single cell level.
NOMe-seq (Nucleosome Occupancy and Methylome sequencing) is a high-resolution single-molecule nucleosome mapping assay based on the ability of the GpC methyltransferase M.CviPI to methylate nucleosome-free regions. Thus, regions occupied by nucleosomes or regulatory proteins (e.g., transcription factors) are not methylated. Subsequent isolation of DNA, bisulfite treatment, and sequencing can map both protein-occupied and unoccupied regions. In addition, the internal CpG methylation information of the DNA can also be read out.
Please find more information addressing the ATAC-seq method, biological questions and published articles here.
Please find more information addressing the NOMe-seq method, biological questions and published articles here.
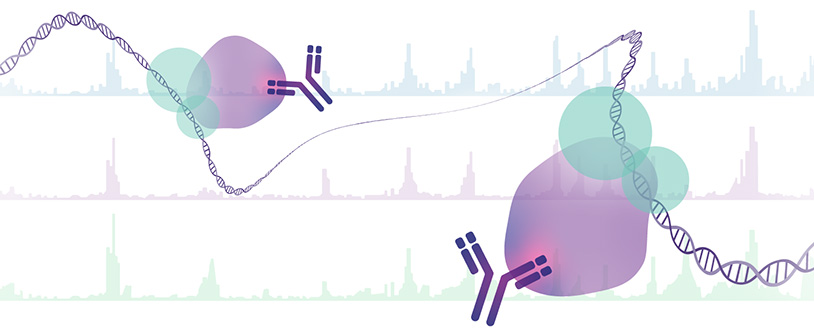
ChIP-seq
The degree of chromatin opening is crucial for the binding of regulatory proteins such as transcription factors. Gene regulatory units such as promoters or enhancers carry characteristic modifications on histone 3 side chains, e.g. methylation at lysine 4 (H3K4me1 and H3K4me3) or acetylation at lysine 9 and 27 (H3K9ac and H3K27ac). During cell aging but also during the development of diseases such as cancer, these modifications change, leading to altered accessibility and thus to altered expression patterns and possibly genomic instability. Specific antibodies can be used to precipitate histone modifications (chromatin immunoprecipitation, ChIP) and isolate and sequence the associated DNA. This enables the generation of genome-wide maps to determine the location and enrichment of specific modifications. Transcription factors bound to DNA can also be mapped in this way.
Please find more information addressing the ChIP-seq method, biological questions and published articles here.
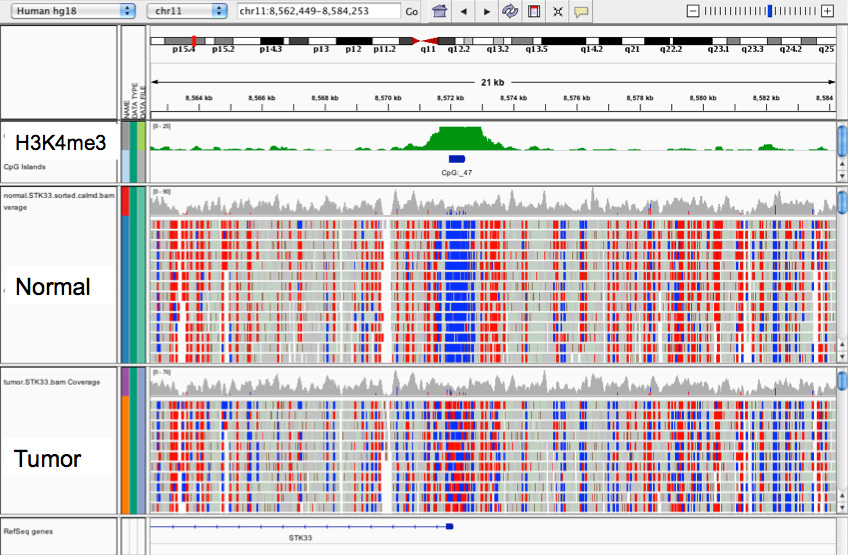
DNA methylation: WGBS and RRBS
Whole-genome bisulfite sequencing (WGBS) enables methylation mapping of whole genomes from tissue, cell cultures, or small cell populations (from 10 ng DNA, approx. 1,500 cells). In this process, isolated chromosomal DNA is treated with sodium meta-bisulfite, which converts unmethylated cytosines to uracils - methylated cytosines are not altered. After ligation of the sequencing adaptors and amplification, the uracils appear as thymins and can thus be distinguished from methylated cytosines by sequencing. Thus, a whole-genome methylation map is obtained, the sequencing effort and costs are comparatively high (about 2,000€ per genome). Please find more information here.
Reduced-Representation-Bisulfite-Sequencing (RRBS) is an adapted variant of Whole-Genome-Bisulfite-Sequencing. The idea is that regulatory regions of the genome such as promoters or enhancers are enriched or highly repetitive, heterochromatic regions are depleted by means of restriction digestion. Thus, only one fraction of the genome is sequenced, which significantly reduces the sequencing effort and cost (about 300€ per sample) without losing valuable information on differential methylation. Please find more information here.

DNA methylation: Hairpin-Bisulfite-Sequencing
The bisulfite treatment of DNA causes the two DNA strands to no longer be complementary to each other. Thus, with conventional bisulfite sequencing, the methylation information can only be read from one strand at a time. By ligating a partially double-stranded DNA hairpin to DNA fragmented by restriction enzymes and subsequently attaching the adaptors to the free ends with subsequent amplification, the information of both strands of the respective fragment can be read. In this way, both local and genome-wide hemimethylation (methylation located on only one of the two strands) can be read out. Hairpin-Bisulfite-Seq can be combined with chromatin accessibility (Hairpin-NOMe-Seq) or mapping of oxidative forms of methylcytosine, e.g. 5'-hydroxymethylcytosine (Hairpin-OxBis-Seq).
Please find more information on the method and further references here.

DNA methylation: EPIC-Array
Illumina's Infinium MethylytionEPIC array provides the ability to screen over 935,000 CpG sites in humans and over 285,000 CpG sites in mice with high consistency across many samples. In this regard, the manufacturer selected those CpGs for the array that have predominantly regulatory function (CpGs in promoters, enhancers, repressors, and insulators). The principle is a primer extension assay performed on the array in massive parallel. The fluorescence-based read-outs are subsequently translated into so-called beta (methylation) values. The robustness of the method and the comparatively low cost (about 200 € per sample) speak for the EPIC array as an alternative to RRBS, especially for a larger sample cohort.
Please find more infos on the method here.

DNA methylation: Local Deep Sequencing
To validate differentially methylated regions detected in genome-wide analyses and to analyze methylation profiles in detail, we perform bisulfite-based local deep sequencing. In this process, the target regions are amplified using so-called fusion primers. Fusion primers already contain the adaptor sequences at their 5' end, which are important for cluster formation and sequencing. This is followed by deep sequencing on the Illumina MiSeq. Qualitative and quantitative analysis of the data is performed using in-house developed R-based pipelines.
Please find more infos on the method here.

Sequencing of bacterial and viral genomes
Microbiomes have recently gained increasing attention. The composition of the gut microbiome, for example, not only plays a major role in gut health and tolerability of dietary components, it is also increasingly associated with neurodegenerative diseases. The diversity of the microbiome can be determined by whole-genome sequencing of bacteria or by sequencing the species-specific variable domains of 16s-rRNA. Both strategies are used in our technical portfolio.
The Corona pandemic continues to pose major challenges to public health systems worldwide. In this context, it is essential to monitor the emergence and spread of SARS-CoV2 virus variants, especially if they lead to altered spread rates and disease severity. We are part of the national COVID19 consortium DeCOI, which has contributed significantly to the understanding of infection, disease and epidemiology through increased sequencing efforts. In addition, the working group is involved in studies investigating the impact of infection on the patient in terms of short- and longer-term epigenetic changes.