
Anwendungen
RNA-Sequenzierung
Im Bereich RNA-Sequenzierung finden vielfältige Fragestellungen Anwendung. Sowohl Gesamt-RNA mit hohem Anteil an ribosomaler RNA als auch angereicherte Formen wie mRNA, kleine (miRNAs) und nicht-kodierende RNAs können mit standardisierten Protokollen sequenziert werden. Neben der Sequenzierung von ganzen Zellverbänden oder Geweben können inzwischen auch Transkriptome von Einzelzellen erstellt werden. Dies trägt zum Verständnis von transkriptioneller Heterogenität zwischen Zellen des gleichen Typus und zwischen Zellen unterschiedlicher Herkunft und Funktion bei.
Umfangreiche Infos zur Methodik, biologischen Fragestellungen und veröffentlichte Artikel finden Sie hier.
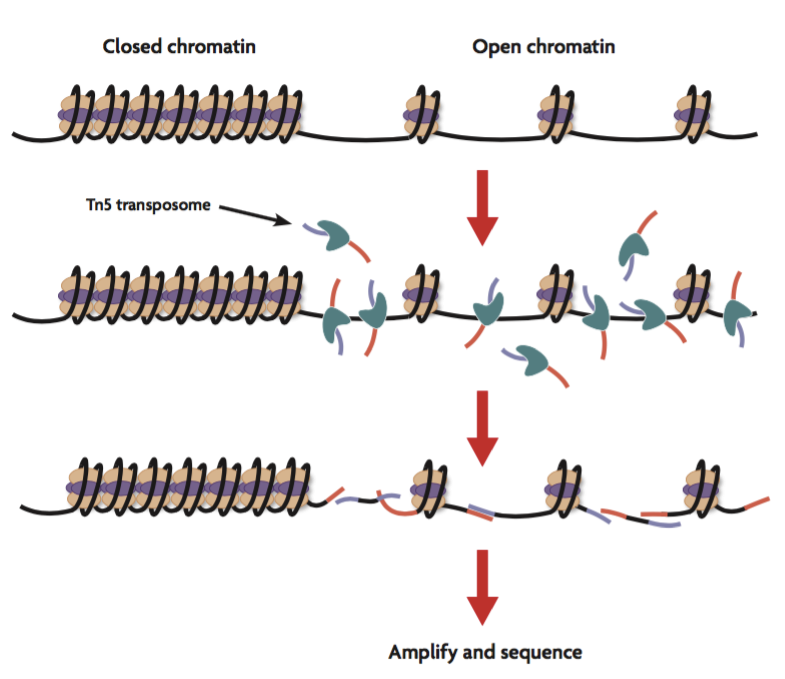
ATAC-Seq/NOMe-Seq
Die Chromatinstruktur hat maßgeblichen Einfluß auf die Genomintegrität und die Steuerung von Genexpression und ist somit ein Schlüsselelement in zellulären Prozessen wie Differenzierung, Alterung und Entartung (z.B. Krebszellentstehung und Progression). ATAC-Seq (Assay for Transposase-Accessible Chromatin sequencing) ist eine Methode zur Kartierung offener Chromatinbereiche, die sowohl für ganze Zellverbände oder Geweben wie auch auf der Einzelzellebene Anwendung findet.
NOMe-Seq (Nucleosome Occupancy and Methylome sequencing) ist eine hochauflösende Einzel-Molekül-Nukleosom-Kartierungs-Assay, der auf der Fähigkeit der GpC-Methyltransferase M.CviPI basiert, Nukleosom-freie Bereiche aufzumethylieren. Damit werden Bereiche, die durch Nukleosome oder regulatorische Proteine (z.B. Transkriptionsfaktoren) besetzt sind, nicht aufmethyliert. Durch anschließende Isolierung der DNA, Bisulfit-Behandlung und Sequenzierung können sowohl Protein-besetzte als auch nicht besetzte kartiert werden. Zudem kann die interne CpG-Methylierungsinformation der DNA mit ausgelesen werden.
Umfangreiche Infos zur ATAC-Seq-Methodik, biologischen Fragestellungen und veröffentlichte Artikel finden Sie hier.
Umfangreiche Infos zur NOMe-Seq-Methodik, biologischen Fragestellungen und veröffentlichte Artikel finden Sie hier.
ChIP-Seq
Der Öffnungsgrad des Chromatins ist entscheidend für das Binden regulatorischer Proteine wie z.B. Transkriptionsfaktoren. Genregulatorische Einheiten wie Promotoren oder Enhancer tragen dabei charakteristische Modifikationen an Histon 3-Seitenketten, z.B. Methylierung an Lysin 4 (H3K4me1 und H3K4me3) oder Acetylierung an Lysin 9 und 27 (H3K9ac und H3K27ac). Während der Zellalterung aber auch bei der Entstehung von Erkrankungen wie Krebs verändern sich diese Modifikationen und führen so zu einer veränderten Zugänglichkeit und damit zu veränderten Expressionsmustern und ggf. genomischer Instabilität. Mit Hilfe von spezifischen Antikörpern kann man Histonmodifikationen ausfällen (Chromatin-Immuno-Präzipitation, ChIP) und die damit verbundene DNA isolieren und sequenzieren. So ist eine Erstellung genomweiter Karten zur Bestimmung von Ort und Anreicherung bestimmter Modifikationen möglich. Auch an DNA gebundene Transkriptionsfaktoren lassen sich so kartieren.
Umfangreiche Infos zur ChIP-Seq-Methodik, biologischen Fragestellungen und veröffentlichte Artikel finden Sie hier.
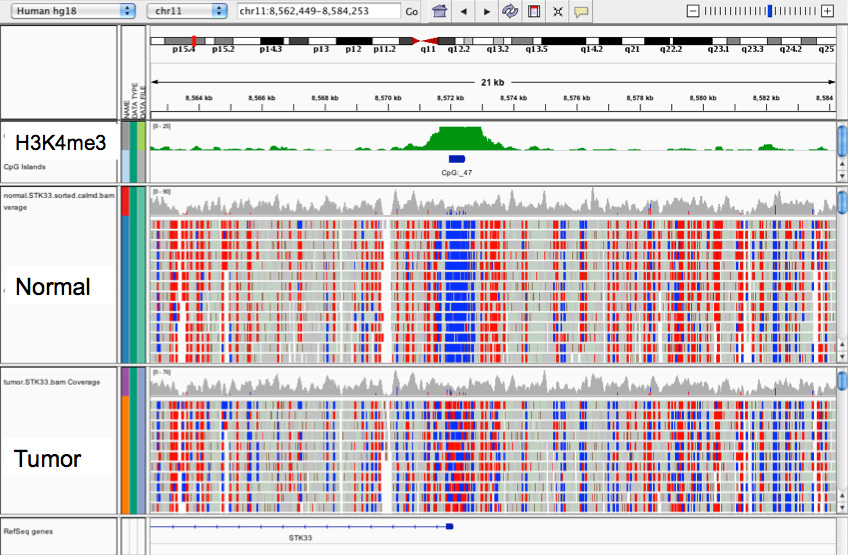
DNA-Methylierung: WGBS und RRBS
Whole-Genome-Bisulfite-Sequencing (WGBS) ermöglicht die Methylierungskartierung ganzer Genome aus Gewebe, Zellkulturen oder geringen Zellpopulationen (ab 10 ng DNA, ca. 1.500 Zellen). Dabei wird die isolierte chromosomale DNA mit Natrium-meta-bisulfit behandelt, wodurch unmethylierte Cytosine in Uracile umgewandelt werden - methylierte Cytosine werden nicht verändert. Nach Ligation der Sequenzier-Adaptoren und Amplifikation erscheinen die Uracile als Thymine und können somit mittel Sequenzierung von methylierten Cytosinen unterschieden werden. Somit erhält man eine gesamtgenomische Methylierungskarte, der Sequenzieraufwand und die Kosten sind vergleichsweise hoch (ca. 2.000 € pro Genom). Weitere Infos finden Sie hier.
Reduced-Representation-Bisulfite-Sequencing (RRBS) ist eine angepasste Variante des Whole-Genome-Bisulfte-Sequencing. Idee ist, daß mittels Restriktionsverdau regulatorische Bereiche des Genoms wie Promotoren oder Enhancer angereichert bzw. hochrepetitive, heterochromatische Regionen abgereichert werden. Somit wird nur eine Fraktion des Genoms sequenziert, was den Sequenzieraufwand und die Kosten erheblich reduziert (ca. 300 € pro Probe) ohne wertvolle Information zu differentieller Methylierung zu verlieren. Weitere Infos finden Sie hier.

DNA-Methylierung: Hairpin-Bisulfite-Sequencing
Die Bisulfit-Behandlung der DNA bewirkt, daß die beiden DNA-Stränge nicht mehr komplementär zueinander sind. Somit kann mit herkömmlichem Bisulfite-Sequencing die Methylierungsinformation immer nur von einem Strang gelesen werden. Durch die Ligation einer partiell doppelsträngigen DNA-Haarnadel (Hairpin) an mittels Restriktionsenzymen fragmentierte DNA und anschließende Anheftung der Adaptoren an die freien Enden mit anschließender Amplifikation kann die Information beider Stränge des jeweiligen Fragments ausgelesen werden. Auf diese Art und Weise kann sowohl lokale wie auch genomweite Hemimethylierung (Methylierung, die sich nur auf einem der beiden Stränge befindet) ausgelesen werden. Hairpin-Bisulfite-Seq lässt sich mit Chromatinzugänglichkeit (Hairpin-NOMe-Seq) oder der Kartierung oxidativer Formen des Methylcytosins, z.B. 5´-Hydroxymethylcytosin (Hairpin-OxBis-Seq) verbinden.
Informationen zu Methodik und Referenzen finden Sie hier.

DNA-Methylierung: EPIC-Array
Der Infinium MethylytionEPIC-Array von Illumina bietet die Möglichkeit, über 935.000 CpG-Stellen im Menschen und über 285.000 CpG-Stellen in der Maus mit hoher Konsistenz über viele Proben hinweg zu untersuchen. Dabei wurden vom Hersteller solche CpGs für den Array ausgewählt, die überwiegend regulatorische Funktion aufweisen (CpGs in Promotoren, Enhancern, Repressoren und Insulatoren). Das Prinzip ist ein Primer-Extension-Assay, der auf dem Array massiv parallel durchgeführt wird. Die Fluoreszenz-basierten Read-Outs werden anschließend in sog. Beta-(Methylierungs-)Werte übersetzt. Die Robustheit der Methode und die vergleichsweise geringen Kosten (ca. 200 € pro Probe) sprechen für den EPIC-Array als Alternative zu RRBS, insbesondere bei einer größeren Probenkohorte.
Infos zur Methodik finden Sie hier.

DNA-Methylierung: Lokale Tiefensequenzierung
Zur Validierung von in genomweiten Analysen detektierten differentiell methylierten Bereichen und zur detaillierten Analyse von Methylierungsprofilen führen wir bisulfit-basierte lokale Tiefensequenzierungen durch. Dabei werden die Zielregionen mit Hilfe von sog. Fusionsprimern amplifiziert. Fusionsprimer enthalten an ihrem 5´-Ende bereits die zur Clusterbildung und Sequenzierung wichtigen Adaptorsequenzen. Es folgt die Tiefensequenzierung auf dem Illumina MiSeq. Die qualitative und quantitative Auswertung der Daten erfolgt durch in-house entwickelte R-basierte Pipelines.
Infos zur Methodik finden Sie hier.

Sequenzierung von Bakterien- und Virusgenomen
Mikrobiome haben in letzter Zeit zunehmend an Aufmerksamkeit gewonnen. Die Zusammensetzung des Mikrobioms im Darm beispielsweise spielt nicht nur eine große Rolle für die Darmgesundheit und Verträglichkeit von Bestandteilen des Nahrungsbreis, es wird auch zunehmend mit neurodegenerativen Erkrankungen in Verbindung gebracht. Die Diversität des Mikrobioms läßt sich durch eine gesamtgenomische Sequenzierung der Bakterien ermitteln oder durch Sequenzierung der Art-spezifischen variablen Domänen der 16s-rRNA. Beide Strategien finden in unserem technischen Portfolio Anwendung.
Die Corona-Pandemie stellt nach wie vor weltweit die staatlichen Gesundheitssysteme vor große Herausforderungen. In diesem Zusammenhang ist es essentiell, die Entstehung und Verbreitung von SARS-CoV2-Virusvarianten zu monitoren, insb. wenn sie zu veränderter Verbreitungsgeschwindigkeit und Erkrankungsschwere führen. Wir sind Teil des nationalen COVID19-Konsortiums DeCOI, das durch verstärkte Sequenzieranstrengungen maßgblich zum Verständnis von Infektion, Erkrankung und Epidemiologie beigetragen hat. Zudem ist der Arbeitskreis an Studien beteiligt, die den Einfluß einer Infektion auf den Patienten oder die Patientin im Hinblick auf kurz- und längerfristige epigenetische Veränderungen untersuchen.